In regulated industries including biotechnology, medical devices, and pharmaceuticals, deviations and Corrective and Preventive Actions (CAPA) are crucial parts of any quality management system. The aforementioned methods are designed to detect, look into, and address any variations from the norm. Deviation and CAPA management, though crucial, can nevertheless be difficult for organizations to manage, frequently resulting in delays, higher expenses, and possible compliance problems. This article will examine typical errors and inadequacies in the examination and handling of deviations and offer helpful advice for efficient handling of deviations and CAPAs. Laboratories may enhance their deviation management procedures and uphold a high standard of quality and compliance in their operations by recognizing and correcting these errors.
Faults in investigations:
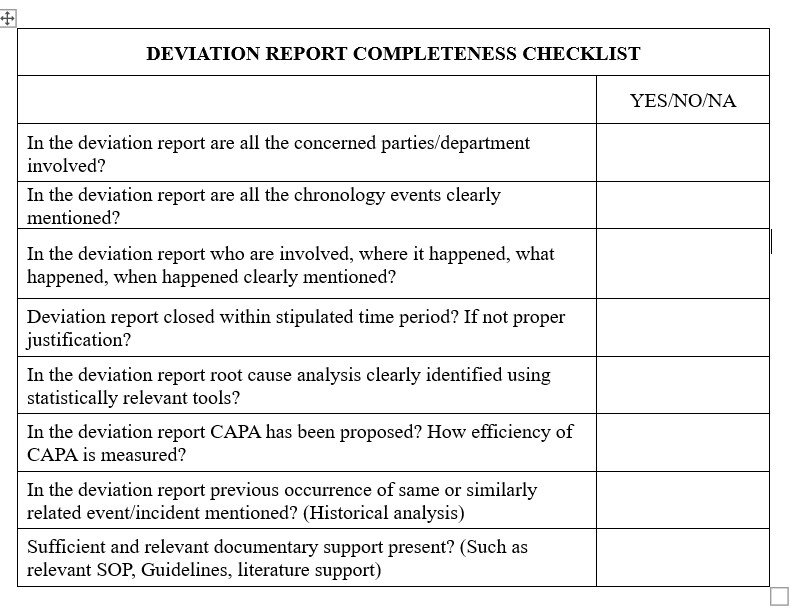
Root causes and their contributory factors were not always fully investigated. There are numerous reasons where the root cause is not obvious. A few primary reasons could be insufficient time and funding, improper investigator training, and a lack of technical and knowledge skill sets.
Significant lack of detail in investigations:
Lack of proper documentation procedure in the investigations.
The information curated over the long history of the organization through investigations contains a wealth of data, which can be utilized for simultaneous improvement, expanding productivity, and reducing the repetitions of errors. Monitoring investigation data will help in understanding types of incidents such as OOS and OOT and their underlying root causes in your organization. Based on the developed and curated data, we can classify the incidents/events and generate actionable insights.
Lack of proper use of tools such as 5-why’s, fish bone analysis.
A reasonable and scientifically proven RCA (root cause analysis) tool should be considered to understand the underlying reason. Further root cause analysis tools such as fishbone analysis, 5 Whys, Kepner-Tregoe, or IS-IS NOT analysis can often tease out a challenging, most probable root cause from an array of discordant facts.
Deviation investigations did not include in-depth level of scrutiny and was not supported by all relevant information.
In numerous cases, proposed changes are not appropriately and completely studied for the risk. Quality Assurance department should consistently access changes to decide their expected impact on the following processes
1) its effect on well established validated process and validated system,
2) Secondly its effect on critical instruments used in production, quality and its related departments.
3) Its effect on product quality, and 4) Finally on its effect regulatory impact.
Faults in execution of Deviation management
No process for measuring the effectiveness of CAPA in line with Quality Risk Management principles. Further quality impact of the CAPAs implemented and deviations taken studies are not carefully assessed. Organizations should verify that change-control was implemented as originally approved, provided the intended output, and has not caused other “undesired” changes. Just like the initiation of change; it should closed in a timely manner.
No reliable measures were taken to prevent reoccurrence (preventive mechanism) is not established in CAPAs.
Effectiveness of change should be measured holistically at all levels. Even after implementation of CAPAs, errors or quality issues re-occur, it shows that Root Cause Analysis is not appropriate, execution mechanism is cumbersome. Therefore CAPAs efficiency should be monitored by Quality Assurance department and appropriate actions to be taken.
Now that you know this hopefully you are in position to answer the Assignment(s)
Are OOT/OOS/OOC/Incidents routinely monitored by your QA department? Do they necessitate conversations and insights with the QC and analytical departments?
What proportions of OOS and events in CAPA are attributable to human error? Does this proportion change yearly or does it decrease?
Does your QA/QC department thoroughly examine root cause analysis using techniques like 5-whys? Does the staff have enough background knowledge to look into root cause analysis?
Does your QA/QC division keep an adequate eye on CAPA effectiveness? How are they quantified?